
Auswirkungen der EU-Medizinprodukte-Verordnung (MDR)
Die Medical Device Regulation, kurz MDR, ist seit 25. Mai 2017 in Kraft und soll die Patientensicherheit deutlich verbessern. Die Verordnung sieht strengere klinische Bewertungen von Medizinprodukten sowie Maßnahmen zur europaweit einheitlichen Benennung und Überwachung von Medizinprodukten vor. Diese Regelungen stellen jedoch insbesondere für KMU einen hohen finanziellen und zeitlichen Mehraufwand dar. Es besteht die Gefahr, dass innovative Entwicklungen kleinerer Unternehmen aufgrund dieser Verordnung möglicherweise gar nicht erst zur Marktreife gelangen.
Hintergrund: Implantat-Skandal 2010
Größtmögliche Sicherheit bei Medizinprodukten, das wünscht sich wohl jeder Patient. Nach dem Skandal eines französischen Brustimplantatherstellers im Jahr 2010 wurde die Qualitätskontrolle von Medizinprodukten und Implantaten auf den Prüfstand gestellt. Damals waren illegal auf den Markt gebrachte Brustimplantate weltweit bei rund 500.000 Frauen eingesetzt worden – mit schwerwiegenden gesundheitlichen Folgen. Doch lange Prüfzeiten könnten nicht nur zu mehr Sicherheit, sondern auch zu Engpässen in der Verfügbarkeit von Medizinprodukten führen. Viele KMUs in der Medizintechnikbranche sehen sich durch die neue EU-Verordnung in ihrer Existenz bedroht.
Verschärfte Vorschriften: Wesentliche Änderungen der Medical Device Regulation (MDR)
Die europäische Medical Device Regulation (MDR) ersetzt die Medizinprodukterichtlinie 93/42/EWG sowie die Richtlinie über aktive implantierbare medizinische Geräte 90/385/EWG und gilt für alle Medizinprodukte mit Ausnahme von In-vitro-Diagnostika.
Die neue Verordnung verschärft nicht nur die allgemeinen Vorschriften für das Inverkehrbringen von Medizinprodukten, sondern auch für die anschließende Marktüberwachung. Sie enthält auch geänderte Anforderungen an die Verantwortung des Herstellers hinsichtlich der technischen Dokumentation, der klinischen Bewertung und der Nachbeobachtung von Medizinprodukten.
Folgende Regelungen sollen zur Verbesserung von Gesundheit und Sicherheit der Patienten und Anwender beitragen:
-
Klassifizierung von Medizinprodukten in Risikoklassen: alle Medizinprodukte werden neu bewertet. Das heißt, die Hersteller müssen die bisherige Klassifizierung gemäß der neuen Verordnung überprüfen und evtl. anpassen. Dies kann auch zu einer Höherklassifizierung von Medizinprodukten führen.
-
Bisherige Konformitätserklärungen und EG-Zertifikate verlieren ihre Gültigkeit. Die Medizinprodukte müssen erneut durch ein Konformitätsbewertungsverfahren zertifiziert werden, um eine neue Konformitätserklärung zu erhalten.
-
Für implantierbare Produkte der Klasse III und aktive Produkte der Klasse II b, die Arzneimittel an den Körper abgeben und/oder aus dem Körper entfernen, gibt es neben dem Konformitätsbewertungsverfahren ein zusätzliches Prüfverfahren, das so genannte Scrutiny-Verfahren.
-
Die Technische Dokumentation wird zum zentralen Thema: Hersteller müssen alle bisherigen Technischen Dokumentationen überarbeiten. Hinzu kommt, dass klinische Bewertungen neu erstellt werden müssen. Sind dafür nicht entsprechende Mitarbeiter vorhanden, müssen ggf. neue eingestellt werden oder externe Mitarbeiter beauftragt werden.
-
Zur Gewährleistung von Transparenz und Rückverfolgbarkeit erhält jedes Produkt eine einheitliche Produktkennzeichnung anhand des weltweiten UDI-Systems (Unique Device Identification System) zur einmaligen Produktkennung.
-
Marktüberwachung: Die Verantwortung des Herstellers nach dem Inverkehrbringen in Bezug auf die Rückverfolgbarkeit von Qualität, Leistung und Sicherheit seiner Medizinprodukte wird neu geregelt.
-
Die MDR Verordnung schreibt unangekündigte Audits vor. Darüber hinaus muss in jeder Organisation eine Person mit der erforderlichen Fachkenntnis für die regulatorischen Anforderungen und deren Einhaltung im Unternehmen verantwortlich sein.
Sicherheitsbedürfnis versus Fortschritt?
Patientensicherheit an erster Stelle
Im Hinblick auf die Patientensicherheit bringt die Medical Device Regulation Verbesserungen, wie die verpflichtende Einführung eines Implantatpasses, die Pflicht zum Abschluss einer Haftpflichtversicherung bzw. zur Bildung ausreichender Rücklagen für Schadensfälle sowie eine Verschärfung des Schutzes von Patientendaten.
…innovative Entwicklungen an zweiter Stelle?
Allerdings stellt sich die Frage, wie sich zusätzliche Sicherheitsmaßnahmen auf die Entwicklung neuer Medizintechnologien auswirken. Aus Sicht der Medizinproduktehersteller bedeutet dies vor allem einen deutlichen Mehraufwand an Zeit, Kosten und Personal. Die Experten des BVMed-Symposiums zum Medizinprodukterecht am 5. Juli 2017 in Köln kamen zu dem Ergebnis, dass die strengeren und aufwändigeren Prüfverfahren dazu führen werden, dass Hersteller künftig mehr Zeit für die Zertifizierung ihrer Produkte einplanen müssen. Zudem wird für die Umsetzung der neuen Anforderungen mehr Personal in der Qualitätssicherung benötigt.
Weniger Ressourcen für Forschung – Engpässe bei der Verfügbarkeit
Durch den erhöhten Zeit- und Kostenaufwand für die Qualitätskontrolle werden in Zukunft zwangsläufig weniger Ressourcen für Forschung und Entwicklung in den Unternehmen zur Verfügung stehen. Dies kann zu Engpässen bei der Verfügbarkeit von Medizinprodukten führen. Insbesondere kleine und mittelständige Unternehmen sehen sich durch die neue Verordnung in ihrer Existenz bedroht.
Höhere Kosten auch für Patienten?
Es stellt sich die Frage, wie die zusätzlichen Kosten finanziert werden sollen. Da Unternehmen den Mehraufwand bei der Herstellung und Zertifizierung ihrer Produkte wieder ausgleichen müssen, könnten sich die Kosten letztendlich auf die Patienten übertragen.
Folge der EU Verordnung: Insgesamt europaweit weniger Benannte Stellen für Medizinprodukte
Auswirkungen hat die MDR auch auf die sogenannten Benannten Stellen. Dabei handelt es sich um neutrale Auditier-, Zertifizier- und Prüfstellen für Produkt- und Qualitätsmanagementprüfungen von Medizinprodukten. Durch die verschärften Auflagen hat ihre Anzahl bereits stark abgenommen, was den organisatorischen Aufwand bei den Herstellern weiter erhöht.
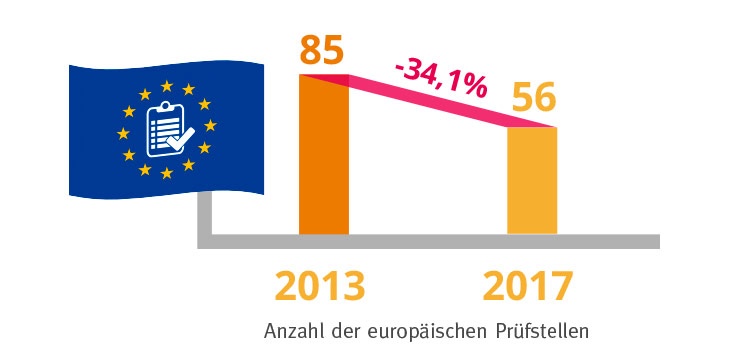
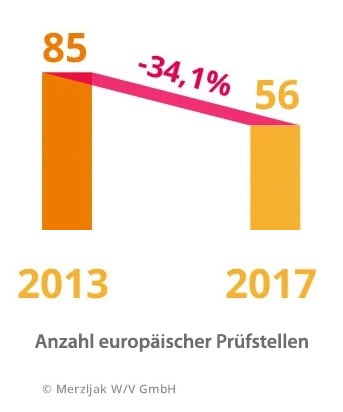
Insgesamt hat sich bereits durch die gestiegenen Anforderungen die europaweite Anzahl der Benannten Stellen seit dem Jahr 2013 von 85 auf 56 verringert. Hier entsteht eine hohe Unsicherheit bei den Medizintechnik Unternehmen, mit welchen Auditierungsstellen sie zusammenarbeiten bzw. ihr Verfahren durchführen können.
Durchführung von Audits im Ausland
Längere Zeitspannen bis zur Zertifizierung von Produkten könnten zukünftig auch durch Auditierungen im Ausland entstehen. Denn die MDR führt auch zu neuen Risikobewertungen von Produkten, die aus dem Ausland importiert oder dort unter eigenem Namen hergestellt werden. Die Kosten für unangekündigte Audits sind laut MDR vertraglich zwischen Hersteller und Benannter Stelle zu vereinbaren. Unangekündigte Audits im außereuropäischen Ausland werden daher möglicherweise teurer ausfallen. Daher bleibt abzuwarten, wie sich die Audit-Anforderungen auf die zukünftige Produktion im Ausland auswirkt, wenn z. B. eine visumpflichtige Einreise dem Charakter eines unangekündigten Audits widerspricht und organisatorische Kosten zu einer Neubewertung der ausländischen Produktion führt.
Hemmschuh für medizintechnische Innovationen
Gerade in Zeiten des demographischen Wandels und fortschreitender Digitalisierung sind oftmals „schnelle“ Entwicklungen gefragt, um konkurrenzfähig zu bleiben und auf gegenwärtige Herausforderungen in der Medizin zu reagieren. Die aufwendigeren Prüfverfahren könnten die technologische Entwicklung in dieser Hinsicht stark hemmen.
Verkürzte Zulassung in Japan
Dass es auch anders geht, zeigt ein Blick nach Japan. Gerade weil Japan sehr stark vom demographischen Wandel betroffen ist, wird hier auf innovative Therapien gesetzt, die möglichst schnell von Patienten genutzt werden können. Um das zu erreichen setzt Japan in bestimmten Bereichen auf die Umsetzung von deutlich verkürzten Zulassungsprozessen bis 2018. So z.B. im Bereich Stammzelltherapien und regenerativer Medizin.
Wie können Medizinprodukte-Hersteller auf die EU Medizinprodukte Verordnung reagieren?
Auch der Bundesverband Medizintechnologie (BVmed) betont, dass die neuen Regelungen eine erhebliche finanzielle und zeitliche Belastung für kleine und mittlere Unternehmen (KMU) darstellen. Die strengeren und aufwändigeren Prüfverfahren führen zu längeren Zertifizierungszeiten und erschweren den Markteintritt neuer Medizinprodukte. Dies kann zu Engpässen in der Verfügbarkeit von Medizinprodukten führen. Der BVmed sieht viele KMU in der Medizintechnikbranche durch die MDR in ihrer Existenz bedroht und plädiert dafür, die Belastungen für KMU zu reduzieren und Innovationen in der Medizintechnik weiter zu fördern.
Effizientes Healthcare Marketing als mögliche Strategie
Insbesondere in Zeiten des digitalen Wandels im Gesundheitswesen, der neuen Regelungen der europäischen Medical Device Regulation (MDR) und immer besser informierter Kunden steigen die Anforderungen an die Produktentwicklung und Vermarktung neuer, innovativer eHealth-Medizinprodukte. Dabei gilt es, Produkttrends frühzeitig zu erkennen und im Medizinproduktemarketing den Informationsbedürfnissen der Kunden in jeder Phase der Customer Journey gerecht zu werden.
1. Online-Auftritt: übersichtlich und informativ
Überzeugen Sie mit Ihren Produkten durch einen professionellen Online-Auftritt. Gerade bei einer Vielzahl von Auswahlmöglichkeiten benötigen Kunden Orientierung und fundierte Produktinformationen. Neben spezifischen Informationen zu Produkt, Finanzierung und Service sollten Ihre Website und alle weiteren digitalen Kanäle übersichtlich und klar strukturiert sein.
2. Kaufentscheidungen aktiv unterstützen
Je besser Sie Ihre Kunden kennen, desto besser wissen Sie, welche neuen innovativen Produkte in Zukunft interessant sein werden. Die Auswertung Ihrer Website kann hier wertvolle Informationen liefern. Mit Digital Marketing Tools können Sie die Performance einzelner Inhalte auf Ihrer Website auswerten. So können Produkttrends frühzeitig erkannt und analysiert werden.
3. Gezielte Kundenansprache und Kundenpflege
Nichts ärgert potenzielle Kunden mehr als falsche Informationen. Sei es durch Unwissenheit oder fehlende Möglichkeiten der nutzerbasierten Datenanalyse. In Verbindung mit einer Marketing Automation Plattform haben Sie jederzeit Zugriff auf alle wichtigen Informationen. Durch fundierte Informationen und Zuverlässigkeit können Sie besser auf die Wünsche Ihrer Zielgruppe eingehen und letztendlich mit Ihren Produkten überzeugen.
Begegnen Sie den Herausforderungen der MDR mit gezielten Healthcare Marketing Strategien!
Auch wenn Veränderungen im Markt durch die neue EU-Verordnung nicht aufzuhalten sind, gilt es für Großkonzerne ebenso wie für kleine und mittelständige Unternehmen, die Herausforderungen bei der Entwicklung, Produktion und im Vertrieb von Medizinprodukten auch künftig zu meistern.
Ob Für Innovationen, bestehende Medizinprodukte oder Ihre Dienstleistungen: Mit Qualität statt Quantität können Sie Ihre Kunden langfristig überzeugen. Marketing Automation mit einer Softwareplattform hilft Ihnen, den Informationsbedürfnissen Ihrer Kunden gerecht zu werden und Ihre Produkte optimal zu vermarkten. Sie sparen Zeit, arbeiten effizienter und stärken die Kundenbindung.
Schließlich geht es langfristig auch um die Zukunft der Medizinprodukte-Industrie und den Medizinproduktemarkt in Deutschland.

Quellen:
http://www.goingpublic.de/wie-innovationsfreundlich-ist-der-gesundheitsmarkt
https://www.bvmed.de/de/bvmed/presse/pressemeldungen/bvmed-zur-neuen-eu-medizinprodukte-verordnung
https://www.bvmed.de/de/bvmed/publikationen/bvmed-newsletter/bvmed-newsletter-27-17/eu-medizinprodukte-verordnung-mdr-aufwand-und-kosten-steigen-fuer-die-hersteller-sorge-vor-engpaessen-bei-den-benannten-stellen
https://idw-online.de/de/news678893
https://www.nzz.ch/meinung/auswirkungen-der-neuen-medizintechnik-regulierung-der-eu-wenn-die-politik-ein-monster-gebiert-ld.153215
https://mednic.de/eu-medizinprodukteverordnung-fordert-die-3d-druckbranche/5002
http://www.tuev-sued.de/akademie-de/seminare-gesundheitswesen-und-medizintechnik/medizintechnik/medical-device-regulation-mdr
http://www.devicemed.de/mdr-die-wichtigsten-neuerungen-im-ueberblick-a-586093/
http://eur-lex.europa.eu/legal-content/DE/TXT/HTML/?uri=OJ:L:2017:117:FULL&from=DE
http://derstandard.at/2000044822799/Medizinprodukte-Schlechtere-Versorgung-durch-ueberzogenes-Sicherheitsdenken
https://www.emergogroup.com/de/blog/2014/03/japanische-behoerden-planen-beschleunigung-der-zulassungsverfahren-fuer-medizinprodukte
http://www.medcert.de/wp-content/uploads/Unangekuendigte-Audits-Bundesanzeiger-2016.06.13.pdf
https://www.medizintechnologie.de/aktuelles/nachrichten/2017-1/europa-zieht-an-den-usa-vorbei/
https://www.medizintechnologie.de/aktuelles/nachrichten/2016-1/medizintechnik-boomt-weltweit/